
Bayesian Analysis for Epidemiologists Part IV: Meta-Analysis
Introduction: Meta-analysis of Magnesium clinical trials
We’ll pick up from the previous section on hierarchical modeling with Bayesian meta-analysis, which lends itself naturally to a hierarchical formulation, with each study an “exchangeable” unit. Let’s first go through a quick illustration of a Bayesian meta-analysis. Don’t worry if it doesn’t entirely make sense right away (though if it does, kudos). The rest of the material on this page goes into details and explains how to conceptualize and code a Bayesian meta-analysis. Much of this material comes from a workshop run by Keith Abrams in Boston in the spring of 2008.
But first, note that I am not here addressing the critically important issues of searching for and evaluating the studies that go into a meta-analysis, or even the (sometimes) contentious issue of meta-analysis itself. For that, take a look at this set of notes
Consider data from 8 RCT’s of magnesium sulfate for myocardial infarction. Mortality ranges from 1% to 17% with sample sizes from 50 to more than 2,300. We assume a normal approximation to calculate a log odds ratio (\(y_j\)) and standard error (\(s_j\)) for each study.
| Magnesium | Placebo | |||||
|---|---|---|---|---|---|---|
| Study | Deaths | Patients | Deaths | Patients | Log OR | SD |
| Morton | 1 | 40 | 2 | 36 | -0.83 | 1.25 |
| Rasmussen | 9 | 135 | 23 | 135 | -1.06 | 0.41 |
| Smith | 2 | 200 | 7 | 200 | -1.28 | 0.81 |
| Abraham | 1 | 48 | 1 | 46 | -0.04 | 1.43 |
| Feldstedt | 10 | 150 | 8 | 148 | 0.22 | 0.49 |
| Schechter | 1 | 59 | 9 | 56 | -2.41 | 1.07 |
| Ceremuzynski | 1 | 25 | 3 | 23 | -1.28 | 1.19 |
| LIMIT-2 | 90 | 1159 | 118 | 1157 | -0.30 | 0.15 |
The first level of the hierarchical model assumes \({y}_{j} \sim {\text{N}}({\theta}_{j},{\sigma}^{2}_{j})\)
As we have noted a few times already, we start by examining 3 possible assumptions about the \({\theta}_{j}'s\):
- We may believe there is no heterogeneity among the studies. So \({\theta}_{j} = \mu\) for all \(j\) Basically, every data point is a realization of a single homogenous study.
- Or, there is complete heterogeneity. There is a different and unrelated \({\theta}_{j}\) for each study, each with its own \(\mu\). This is a group of 8 unrelated studies.
- Or, that the studies are exchangeable. We have seen this is a compromise between complete homogeneity and complete heterogeneity. The studies are not identical, but they are related in a way that knowing something about, say, 7 studies will tell us something about the 8th study.
I haven’t emphasized this before, but exchangeability is not a trivial assumption and requires careful thought. In this particular case, we might be concerned that there is some systematic difference among the study sites which we would want to account for in our model, perhaps with a covariate. Or, we may believe there are subgroups that should be modeled separately. If though, we are reasonably confident in our assumption of exchangeability, we may specify a second level of variance as \(\theta_j \sim N(\mu, \tau^2)\) , with the \({\theta}_{j}\)’s drawn from a normal distribution with hyperparameters \(\mu\) and \(\tau^2\), which is the variation in the population-level effects.
As in our previous Bayesian analyses, \(\theta_j\) then is a compromise between the data (\(y_j\)) and the prior. In this case, it can be shown (though not by me) that the the weights provided by the inverse variances of the hyperparameters:
\[\hat{\theta}_{j} = \frac{\frac{1}{{\sigma}^{2}_{j}}{y}_{j} + \frac{1}{{\tau}^{2}}\mu} {\frac{1}{{\sigma}^{2}_{j}} + \frac{1}{{\tau}^{2}}}\hspace{1cm}{\text {and}}\hspace{1cm} V_{j} = \frac{1}{\frac{1}{{\sigma}^{2}_{j}} + \frac{1}{{\tau}^{2}}}.\]
The exchangeable model therefore leads to narrower posterior intervals than the complete heterogeneity model, shrunk toward the prior mean. The degree of shrinkage depends on both the variability between studies (\(\tau^2\)) and the precision of each treatment effect (\(\sigma^2_j\)) In the following image, note that large trials have relatively little shrinkage while smaller trials have more shrinkage.
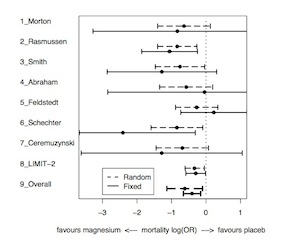
Bayesian Approaches to Meta-Analysis and Data Synthesis
Let’s consider a group of 22 studies that examined the cardiac protective benefits of beta blockers. Using data extracted from the study reports, we can calculate the odds ratio for each study as the classic \(\frac{ad}{bc}\), and then transform each odds ratio to the log scale for 22 outcome variables, \(Y=ln(OR)\) each of which is associated with a variance \(\sigma^2 = V= \frac{1}{a} + \frac{1}{b} + \frac{1}{c} + \frac{1}{d}\)
We can model these data in JAGS with the following code for the likelihood, where (we assume) the log OR’s are normally distributed:
1 2 3 4 5 6 | model {
for (j in 1:Nstud) {
P[j] <- 1/V[j] # precision
Y[j] ~ dnorm(delta[j],P[j])
delta[j] ~ dnorm(d,prec)
} |
We continue the code with the following priors, including a uniform prior on the standard deviation, and calculations for between-study variance (tau.sq), exponentiation of the mean log OR to get an overall effect estimate, and the use of a step function to return the probability that that synthesized OR estimate is greater than 1.
1 2 3 4 5 6 7 | d ~ dnorm(0,1.0E-6) prec <- 1/tau.sq tau.sq <- tau*tau # btw-study var tau ~ dunif(0,10) # Uniform on SD OR<-exp(d) #exponentiate to get back to OR scale prob.OR1<-step(d) #calc prob of OR > 1 } |
A few points about this code. First, we work on the log scale. This stabilizes things, results in better statistical properties and makes it easier to calculate variances for common epidemiologic metrics like the odds ratio (for which, as indicated above, the log OR has a variance of \(\sigma^2 = \frac{1}{a} + \frac{1}{b} + \frac{1}{c} + \frac{1}{d}\)) and the relative risk (for which the variance of the log RR is \(\sigma^2 = \frac{1}{a} + \frac{1}{a+b} + \frac{1}{c} + \frac{1}{c+d}\)).
After the difficult work of identifying and evaluating studies for inclusion in your synthesis, and then extracting the necessary data points to calculate odds ratios for an effect estimate, setting up the data is relatively straightforward. For each study, enter the data into R as an object of your choice (vector, matrix, etc…) so that for each study you can calculate an odds ratio, it’s natural log, and the variance for the log OR. 1
The usual (non-Bayesian) approach to synthesizing the effect estimates is to combine the individual estimates by weighting them by the inverse of their variance. With BUGS or JAGS, we specify prior distributions and use MCMC methods.
Fixed Effects vs. Random Effects
Meta-analyses can be broadly categorized as “fixed effect” or “random effect” models.
Fixed Effect Model
In a fixed effect model, all studies are assumed to be estimating the same underlying effect size “d”, a single parameter that varies randomly, e.g. \(Y_i \sim N(d,V_i)\). We are interested in estimating the parameter “d” to describe the synthesized evidence. In a Bayesian approach, we assign a prior distribution to “d” and combine that with the available data (the study-level estimates) in the form of a likelihood to calculate a posterior distribution from which we will draw inferences. As is always the case, the choice of a prior deserves close attention, particularly if you don’t have a lot of information or data (If there is a lot of data, or information form individual studies, the likelihood will overwhelm any reasonable prior for “d”). One reasonable choice is \(d \sim N(0, 105)\) 2 Which translates to a 95% certainty that true value of d lies between -1.96x316 and +1.96x316 (316 comes from \(\sqrt{105}\)) On an odds ratio scale, this would be a 95% certainty that the odds ratio lies between \(10^{-269}\) and \(10^{269}\). We could choose a flat prior, but would have to explicitly specify start and stop points. We could also alternatively choose an improper prior, but BUGS and JAGS don’t play very nicely with them.
Here is some sample code for such a normally distributed, single parameter, fixed effects meta-analysis:
model
{
for (i in 1:Nstud)
{
P[i] <- 1/V[i]
y[i] ~ dnorm(d, P[i])
}
d ~ dnorm(0, 1.0E-5)
OR <- exp(d)
}
The only initial value we need, is for “d” (which can be set to 0 for initial runs). Note that unlike the previous code, we are not allowing a second level of variation on the mean log-odds ratio. It is, in effect, “fixed”.
Random Effects Model
We might believe that it is unreasonable to assume that all the studies in our meta-analysis are estimating exactly the same treatment effect, and that are they are the only studies in which we are interested, or perhaps the only studies that exist on the topic. We may assume that differences in samples, design, and conduct introduce more statistical heterogeneity between studies, than can reasonably be attributed to random error within studies. In that case it might be more reasonable to use a random effects model. A random effects model assumes the studies are a sample from all possible studies and includes an additional variance component for variation or heterogeneity between studies.
Notice the emphasis on statistical heterogeneity. We are not addressing the important issue of systematic bias or confounding. The additional structured error term of the kind described in random effects models is by no means a panacea. There are critics (notably Sir Richard Peto and his colleagues) of random effects models. Among the arguments are that the approach blindly addresses the additional variance without in any way explaining it. In response, many practitioners argue that while not perfect, it is essentially a more conservative approach than a fixed effect model. If the current literature is any indication, this view is clearly in the majority.
From a Bayesian perspective, a random effects model corresponds to the kinds of hierarchical models we have already considered in a previous section of these notes. Now, in addition to within-study variation, \(Y_i \sim N(d,V_i)\), we have a parameter for between-study variation, \(d_i \sim N(\delta, \tau^2)\). The prior on this variance component (\(\tau^2\)) for between study variation is a bit tricker to navigate. It can’t be allowed to be negative, so a uniform distribution, say, \(\tau^2 \sim U(0,10)\) has been proposed as reasonable. But given the often small number of studies in meta-analyses, the choice of prior can be influential, and this has been an area where there a number of priors have been proposed as perhaps more “reasonable”. You will see some studies using a Gamma(0.001, 0.001) as a precision term, or a N(0,1)[0,1], where the [0,1] notation means it is truncated at 0 so forced to be negative, or a Uniform (1/1000, 4) prior. The more studies in your analysis, the less of a problem this is, with a rule of thumb that somewhere between 20 and 30 studies is a good place to be. With fewer studies, sensitivity analyses using different priors for \(\tau^2\) is strongly recommended. In any event, because it takes the variance component \(\tau^2\) into account, you will find that the credible intervals in a Bayesian approach are often wider than the confidence intervals in a frequentist approach to the same data.
Following is (again) the BUGS/JAGS code for a random effects Bayesian model:
model
{
for (i in 1:Nstud)
{
P[i] <- 1/V[i]
y[i] ~ dnorm(delta[i], P[i]) #study effects
delta[i] ~ dnorm(d, prec) #random level effects between studies
}
d ~ dnorm(0, 1.0E-5)
OR <- exp(d)
tau~dunif(0,10)
tau.sq<-tau*tau
prec<-1/(tau.sq)
p.harm<-1-step(d) # prob OR < 1
}
The last line of code for “p.harm” is a nice way to get the probability of the synthesized estimate being below (or above) some value, and is an example of the kind of think that can only really be done in BUGS or JAGS.
Up to now, we’ve been modeling the odds ratio on the log scale. We can model odds ratios directly, rather than the log ORs. This approach doesn’t carry with it the assumption of normality of the individual estimates, and may be better in the setting of rare events, since on the log scale we have to cheat a bit and add, say, 0.5 to zero observations to get a log transformation. At its most basic level, you will get the same results as the log-normal model above, but it is more generalizeable, and can be built up into a meta-regression.
Following is some BUGS / JAGS code for a direct odds ratio model.
model
{
for( i in 1 : Nstud ) {
rA[i] ~ dbin(pA[i], nA[i])
rB[i] ~ dbin(pB[i], nB[i])
logit(pA[i]) <- mu[i]
logit(pB[i]) <- mu[i] + delta[i]
mu[i] ~ dnorm(0.0,1.0E-5)
delta[i] ~ dnorm(d, prec)
}
OR <- exp(d)
d ~ dnorm(0.0,1.0E-6)
tau~dunif(0,10)
tau.sq<-tau*tau
prec<-1/(tau.sq)
}
it is a binomial model, where:
- nA[i], nB[i] are the total number of patients in the two arms of the study, or the exposed/unexposed populations
- rA[i], rB[i] are the number of events in the two arms
- pA[i], pB[i] are the underlying probabilities used to define the likelihood
- mu[i] is the log odds event in group A (and requires a prior)
- delta[i] is the so-called “treatment effect” or the log OR for the ith study, and
- d is the study-level point estimate, with tau the between study variance
Example: Random Effects Meta-Analysis of Beta Blocker Studies
Beta blockers are a class of drug most often used to treat hypertension. A number of studies have looked at the efficacy of beta blockers in preventing death after a myocardial infarction (heart attack). Let’s synthesize the results of these studies using data for the natural logs of the odds ratios.
First, the model code:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | cat("model { # Likelihood for (j in 1:Nstud) { P[j] <- 1/V[j] # Calculate precision Y[j] ~ dnorm(delta[j],P[j]) delta[j] ~ dnorm(d,prec) } # Priors d ~ dnorm(0,1.0E-6) prec <- 1/tau.sq tau.sq <- tau*tau # tau.sq = between-study variance tau ~ dunif(0,10) # Uniform on SD OR<-exp(d) #exponentiate to get back to OR scale prob.OR1<-step(d) #calc prob of OR > 1 }", file="betaMeta1.txt") |
Next, we read in the data, and set up initial values and nodes to monitor
1 2 3 4 5 6 7 8 9 | blocker<-read.csv("...path to your data.../BLOCKER.csv", sep=",", header=T) Nstud<-22 blockerDat <- list(Y=blocker$Y,V=blocker$V,Nstud=Nstud) blockerParams <- c("d","tau","OR", "prob.OR1") blockerInits<-function(){ list("delta"=c(0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0),"d"=c(0), "tau"=c(1)) } |
Now, run the model in JAGS using R2jags and look at some results:
1 2 3 4 5 6 7 8 9 | set.seed(100) library(R2jags) jag.blocker1<-jags(data=blockerDat, inits=blockerInits, parameters.to.save=blockerParams, n.iter=1000, n.thin=20, model.file="betaMeta1.txt") jag.blocker1 |
Two quick conclusions from these results are that (1), the summary or synthesized estimate for the effect of beta blocker on post-MI mortality is 0.78 with a 95% credible interval of 0.69 to 0.90. Based on the predictive distribution, the probability that a future study will show harm is virtually nil.
As an exercise, try coding this as a binomial model on the OR scale. Instead of using the log data above, use these data (defined as above) and these initial values
1 2 3 4 5 6 7 8 9 10 11 12 13 | list(rA = c(3, 7, 5, 102, 28, 4, 98, 60, 25, 138, 64, 45, 9, 57, 25, 33, 28, 8, 6, 32, 27, 22 ), nA = c(38, 114, 69, 1533, 355, 59, 945, 632, 278,1916, 873, 263, 291, 858, 154, 207, 251, 151, 174, 209, 391, 680), rB = c(3, 14, 11, 127, 27, 6, 152, 48, 37, 188, 52, 47, 16, 45, 31, 38, 12, 6, 3, 40, 43, 39), nB = c(39, 116, 93, 1520, 365, 52, 939, 471, 282, 1921, 583, 266, 293, 883, 147,213, 122, 154, 134, 218, 364, 674), Nstud = 22) list(d = 0,tau=1, mu = c(0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0), delta = c(0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0)) |
Once again, if you must cheat, the answer is here
Heterogeneity and Prediction
A summative measure of the overall effect is nice, but doesn’t tell whether, or how, the effects varied from study to study. Were there, for example, both harmful and beneficial results among the studies. Forest plots are a good way to illustrate effects across studies and should be considered mandatory. In a Bayesian setting, \(\tau^2\) in a hierarchical model, as defined above can be used to assess heterogeneity by allowing us to define a so-called “predictive distribution”. We use the \(\tau^2\) result from the meta-analysis to assess the heterogeneity between studies and define a probability distribution for most likely outcome in a future study. The predictive distribution, \(d_new \sim (d, \tau^2)\) can be easily coded into the model syntax:
d.new<-dnorm(d, prec) OR.new<-exp(d.new)
You could include a step statement to calculate the probability that a future study will demonstrate a beneficial (or deleterious) outcome.
Heterogeneity and Meta-Regression
As noted above, a random effects model can account for increased heterogeneity, but doesn’t explain it. If you are interested in explaining or controlling for the additional heterogeneity, you can conduct stratified analyses, or regressions. Stratified analyses are suited to discrete characteristics or covariates by which you can group your sample of estimates. The approach is relatively straightforward. Stratify your data by study level characteristic(s), e.g. length of follow up, clinic setting, etc…
Regression approaches, sometimes termed “meta-regressions” can accommodate both discrete and continuous characteristics, relating the size of the summary estimate to study-level characteristics like (say) study quality. You can also code the data for summary study-level characteristics, e.g. percent female, and relate that to the outcome summary estimate. The general approach is much like the random effect model we have already seen:
- \(Y_i \sim N(d_i, \beta_{xi}, V_i)\) for the \(i^{th}\) study
- \(d_i \sim N(d, \tau^2)\)
But we are adding \(\beta\) coefficients for the covariate values. We will use a normally distributed, only vaguely informative (i.e. large variance) prior for \(\beta\)
As an example, consider a Bayesian meta-analysis of studies looking at the effectiveness of BCG vaccine in preventing tuberculosis. A \(\tau =0.37\) on the log scale indicates a lot of heterogeneity across studies. Rather than just account for it with a random effects term, the investigators conducted a meta-regression to explain the possible effect of distance from the equator for the study setting. The following graph plots BCG treatment effect on the y axis by distance from the equator on the x axis, with an ab line from a meta-regression.
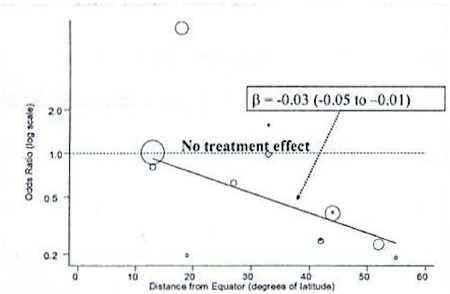
The following, is syntax for such a meta-regression:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 | model
{
for( i in 1 : Nstud ) {
rA[i] ~ dbin(pA[i], nA[i])
rB[i] ~ dbin(pB[i], nB[i])
logit(pA[i]) <- mu[i]
logit(pB[i]) <- mu[i] + delta[i] + lat[i]*beta
mu[i] ~ dnorm(0.0,1.0E-5)
delta[i] ~ dnorm(d, prec)
}
OR <- exp(d)
d ~ dnorm(0.0,1.0E-6)
tau~dunif(0,10)
tau.sq<-tau*tau
prec<-1/(tau.sq)
beta ~ dnorm(0.0,1.0E-6) #additional prior for beta
} |
Note that, really, the only major difference from previous models is the addition of a \(\beta\) coefficient (“beta”) for a study-level variable on latitude (“lat”) for degrees from the equator. We look at this beta coefficient to see if it indicates a meaningful, linear change in the effectiveness the farther you get from the equator. In fact, as indicated in the figure above, there was a meaningful effect, and it was accompanied by a significant decline in heterogeneity as measured by \(\tau\)
As is always the case, some caution is in order. Sub group analyses and meta-regression can be problematic in a number of respects. First, power is based on the number of studies not the number of people in those studies. Second, the studies themselves are often observational, not experimental, and subject to all the issues of bias and confounding to which observational studies are subject. Finally, you are often dealing with average values for study, so there is there are additional issues of potential ecologic fallacy.
Example: Bristol Pediatric Cardiac Mortality Study
We will usually want to compare the individual estimates in a meta-analysis. An example of this comes from the Bristol Pediatric Cardiac Mortality Study. Data were collected on 30-day mortality following pediatric surgery at Bristol Hospital and 11 other UK centers. Following are the data:
| Hospital | Deaths | Surgeries |
|---|---|---|
| 1 | 25 | 187 |
| 2 | 24 | 323 |
| 3 | 23 | 122 |
| 4 | 25 | 164 |
| 5 | 42 | 405 |
| 6 | 24 | 239 |
| 7 | 53 | 482 |
| 8 | 26 | 195 |
| 9 | 25 | 177 |
| 10 | 58 | 581 |
| 11 | 31 | 301 |
| 12 | 41 | 143 |
The following syntax should, by now, be starting to look familiar. We define a binomial likelihood with a log odds of mortality, prior distributions for the nodes, and then set up and run the data structures in R2jags.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 | cat("model{ for( i in 1 : N ) { r[i] ~ dbin(p[i],n[i]) #Likelihood logit(p[i]) <- b[i] #Log-odds of mortality b[i] ~ dnorm(mu,prec) #Random effects model for log-odds mortality } mu ~ dnorm(0.0,1.0E-6) #Priors sd <- 1 / sqrt(prec) prec ~ dgamma(0.001,0.001) }", file="bristol.txt") bristolDat<-list(N=12,r=c(25, 24, 23, 25, 42,24,53,26,25,58,31,41), n=c(187,323,122,164,405,239,482,195,177,581,301,143)) bristolInits<-function(){list(b = c(-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1), prec = 2, mu = -1)} bristolParams<-c("p", "mu", "sd") library(R2jags) jag.bristol<-jags(data=bristolDat, inits=bristolInits, parameters.to.save=bristolParams, n.iter=1000, n.thin=20, model.file="bristol.txt") jag.bristol plot(as.mcmc(jag.bristol)) |
Now, though, rather than having a primary interest in the synthesized or summary measure of effect, we’d like to compare the hospitals. We will use a so-called “caterpillar plot” to do this. We see that one of the small graphics on the default plot method for the jags output produces the kind of information in which we are interested. We could conceivably (though I’ve never been able to) use the str() function to extract the plot in which we are interested. The “mcmcplot” package, though, is tailor made for this task.
1 2 3 4 | plot(jag.bristol) install.packages("mcmcplots") library(mcmcplots) caterplot(as.mcmc(jag.bristol), parms="p") |
We see that hospital 12, appears to be an outlier…
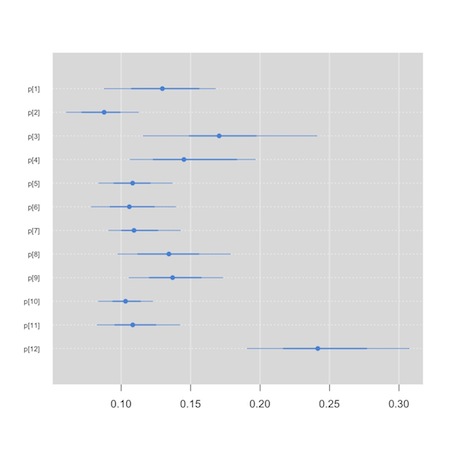
This tool can also be used to create Forest plots for your meta analysis, though there are probably better tools in packages like “meta” and “rmeta” that you can feed your results to. I tend to hand code my Forest plots. Here is a template based on some code Alex Sutton was kind enough to share with me. I used it to create figure 1 in this paper. It is a bit of a hack, but you can adapt it to your needs.3
Your Turn: Back to Meta-analysis of Magnesium clinical trials
At this point, I encourage you to scroll back up to the top of this page and try coding the magnesium trial data for a random effects model using the natural odds ratio scale. The answer is here
There are some R packages, e.g. “meta”, “rmeta”, “metafor”, to make this process and that of creating informative plots even easier.↩
This choice of prior, and as noted above, much of this material, comes from a workshop by Keith Abrams I attended. He has a book on Bayesian approaches to data synthesis. I haven’t read it, but if it is any where near as good as his workshop, it is an easy recommendation.↩
If you come up with something nice, do let me know.↩